Alice Galzignato, Studio Oculistico d’Azeglio, Bologna, Italy
Patient:
A 26-year-old man was referred for a neuro-ophthalmological evaluation following the finding of painless acute visual loss and optic disc edema in his right eye.
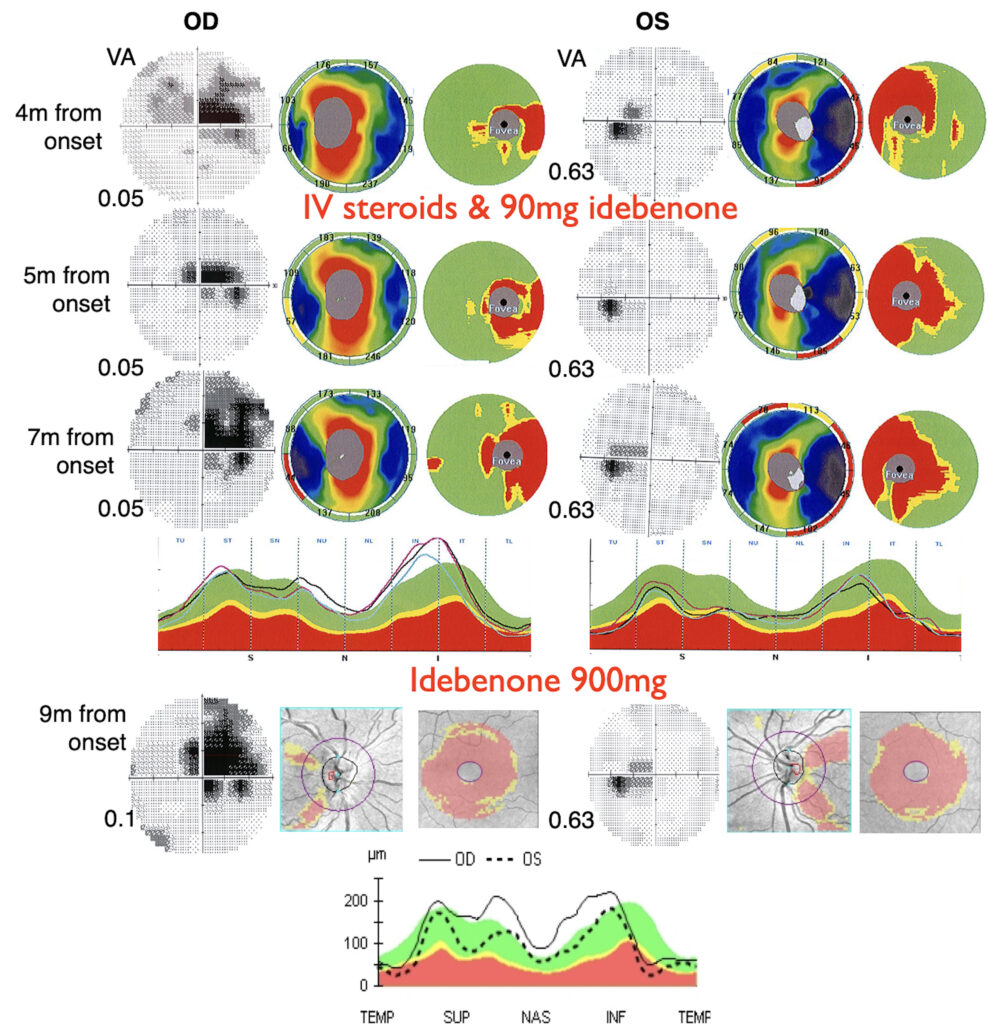
A previous examination four months from onset of right eye visual loss reported a BCVA 0.05 OD and 0.63 OS, color vision defect and central scotoma at VF in OD. The optic disc was described as edematous with telangiectasias OD, and normal OS; whereas, OCT analysis reported RNFL thickening OD. (Figure 1) IV steroid therapy for 5 days was initiated about 6mos. after onset for the presumed diagnosis of optic neuritis. The patient self-administered idebenone 45mg twice. At the one-month follow-up, a slight improvement was noted in visual acuity and visual field.(Figure1). During hospitalization, genetic analysis identified the homoplasmic m.11778G>A mutation in ND4, leading to diagnosis of LHON and an increase in idebenone dosing to 900 mg/day.
In the illustration below and later, use OD and OS instead of RE and LE.
.
Figure 1. Progression of visual field, best corrected visual acuity (VA), retinal nerve fiber layer (RNFL) and ganglion cell layer thickness (GCL) in the right (OD) and left eye (OS) at different timepoints of the follow-up. In the charts of the right eye, a progression of functional and structural damage can be observed over the first nine months, with enlargement of the ceco-central scotoma and progressive thinning of RNFL and GCL. In the left eye, a small ceco-central scotoma and thinning of the temporal RNFL and nasal GCL are evident. At 4 months from onset, 5 days of intravenous steroid and 90mg of idebenone were started, while idebenone 900mg was prescribed at 7 months from the onset.
Neuro-ophthalmological reassessment was made 9 months after presentation. There was a family history of an undiagnosed visual problem in the maternal great-grandmother. The patient had a history of smoking approximately 15 cigarettes per day and engaging in episodic binge drinking. He also reported occasional cocaine use, with the last reported use occurring approximately one month prior to the onset of visual loss. Ophthalmic history reveals left-eye amblyopia and strabismus, treated at the age of 4 with occlusion therapy.
Patient examination showed BCVA 0.1 with Sph +5.25 OD, and 0.63 with Sph +4.25 OS, and alternating esotropia of 10 PD. A slight relative afferent pupillary defect was observed OD. Ishihara test yielded 1/12 plates OD and 11/12 OS, while the HRR test showed 0/6 OD and 4/6 OS.
On fundoscopic examination showed hyperemic optic disc with increased vascular tortuosity in nasal-superior side observed OD, along with temporal pallor of the both optic discs.
OCT scans of the optic nerve head revealed swelling of the nasal and superior RNFL sectors OD, along with temporal RNFL thinning in both eyes and macular GCL thinning bilaterally. (figure 1). The VF test showed a large ceco-central scotoma OD and a small ceco-central scotoma OS.
Over time, the best-corrected visual acuity remained stable at 0.1, whereas visual field testing revealed a progressive enlargement of the ceco-central scotoma. The defect, initially confined to the supero-temporal sector, gradually extended toward the inferior hemifield in an altitudinal pattern, reaching its maximal extent at 18 months from onset, with preservation of the supero-nasal sector.
At OCT analysis, the right optic nerve showed progressive RNFL thinning up to 21 months, after which it remained stable for the following 7 years. GCL thinning progressed until approximately 12 months from onset and then stabilized after 16 months. The left optic nerve exhibited a similar pattern of RNFL and GCL thinning from the first observation; however, compared with the right eye, the left eye retained slightly greater RNFL thickness while showing comparable GCL thinning. (Figure 2, 3).
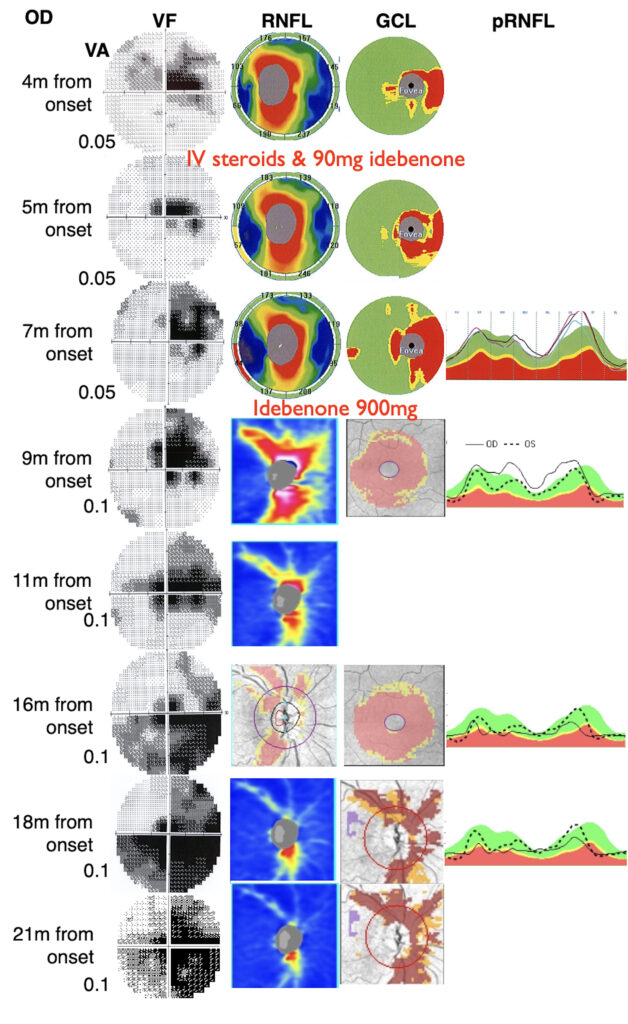
Figure 2. Progression of visual field, best corrected visual acuity (VA), retinal nerve fiber layer (RNFL) and ganglion cell layer thickness (GCL) of the right eye (OD) at different time points of the follow-up. Whenever possible, charts from different imaging devices were collected to document and highlight disease progression. In particular a slowly progression of functional and structural damage was evident; the RNFL thinning progressed up to 21 months and the GCL thinning deepened until approximately 12 months from the onset and then stabilized after 16 months.
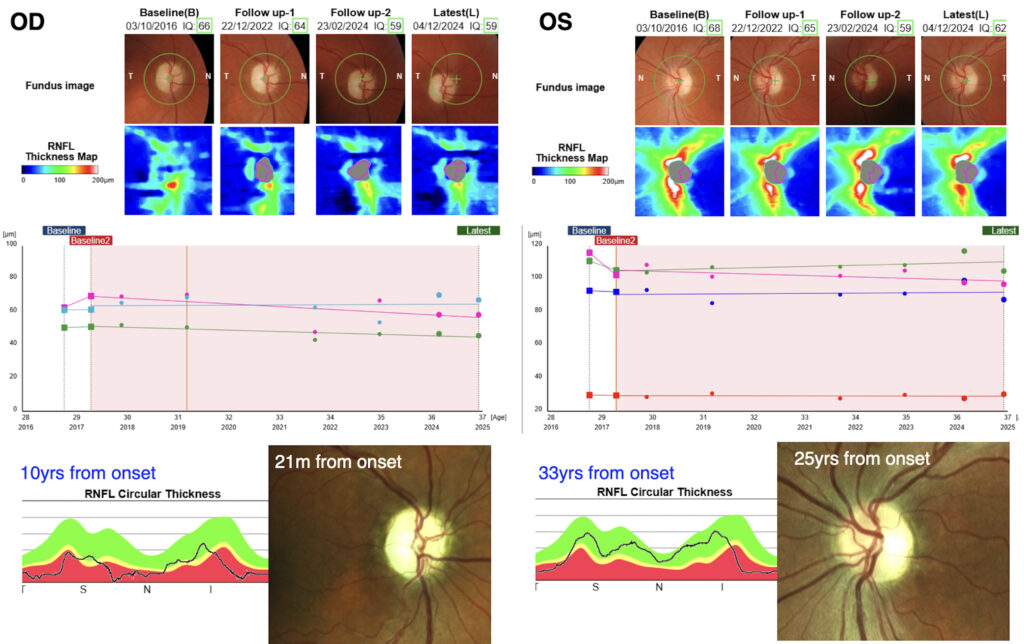
Figure 3. Fundoscopic aspect of the optic nerve head, retinal nerve fiber layer (RNFL) and ganglion cell layer (GCL+) thickness maps of the right (OD) and left (OS) eye at different timepoints (upper panel). The graphs demonstrated a slight RNFL thinning of superior sectors in OD and inferior sector in OS through the follow-up visits from 3 yrs to 10 yrs from onset of OD (middle panel). In the bottom panel are represented the RNFL thickness graph and the optic nerve picture of OD at 10 yrs and 21 months form onset and of the OS at 33yrs and 25yrs from onset respectively.
In view of the temporal pallor of the left optic disc, the reduction in GCL density, along with altered color vision perception and the absence of visual recovery following amblyopia treatment, it was suspected that the initial diagnosis of amblyopia had to be re-considered, and that the patient had instead presented a childhood onset LHON with insidious onset.
From a clinical standpoint, the patient had stopped smoking immediately after the onset of visual decline but resumed smoking as well as alcohol and substance abuse about a year later. Over the following years, he developed a psychotic disorder that required psychiatric hospitalization and treatment with psychotropic medications.
What raises suspicion of a hereditary optic nerve disease?
The main factor that raised the suspicion of a specific hereditary optic neuropathy rather than optic neuritis was the presence of an acute, painless visual loss and optic disc swelling along with the presence of a relative afferent pupillary defect. The ophthalmoscopic appearance of microangiopathy and the swelling of the optic nerve fibers on OCT angiography were indicative of LHON.
Furthermore, the presence of a suspected amblyopia in the fellow eye, associated with thinning of the temporal retinal nerve fiber layer and reduction of macular ganglion cell density; there was no anisometropia or other factors associated with amblyopia, suggested that the left eye may have experienced an early onset of the disease during childhood — a condition often difficult to diagnose due to the subtlety of symptoms in children and the typically more favourable disease course when it begins in early life.
How can amblyopia be distinguished from other causes of visual loss in children?
Distinguishing amblyopia from other causes of visual impairment in children is not always straightforward, due to the difficulty in recognizing symptoms, the variability in disease expression, and the involvement of an immature visual system, especially in cases that remain unilateral for many years.
In a child with suspected amblyopia, it is important not to underestimate the assessment of visual acuity and a comprehensive ophthalmological examination, including color vision, specifically using HRR plates if an optic neuropathy is suspected. Also useful are the fundus examination and pupillary light reflex examination, ideally combined with instrumental evaluations such as OCT and visual field testing. A complete evaluation is particularly important in any amblyopic child who fails to show improvement despite appropriate therapy. Furthermore, strabismus in a child with a positive family history of hereditary optic neuropathy should be considered as a manifestation of LHON.
How to differentiate the various hereditary optic nerve disease?
For the differential diagnosis of hereditary optic neuropathies, a thorough history is essential, focusing primarily on the mode and tempo of of the visual impairment, laterality, and the presence of family history of ocular diseases. Clinical evaluation should then assess the severity of visual loss, the presence of pupillary defects, color vision, and the appearance of the optic nerve head. Instrumental investigations include, first and foremost, optical coherence tomography (OCT) and visual field testing (in children capable of performing it). In this specific case, the presence of swelling of the optic nerve fibers and associated microangiopathy led us to favour a diagnosis LHON. However, for a molecular diagnosis, genetic testing is indispensable.
Discussion
The age of onset in LHON commonly ranges between 15 and 35 years and is usually bilateral, simultaneous or sequential, but atypical clinical presentation include a more variable age of onset, from the first decade to 80 years old, and a slowly progressive clinical course [1-3]. Ref ( Valerio Carelli 1, Pio d’Adamo 2, Maria Lucia Valentino 3, Chiara La Morgia 3, Fred N Ross-Cisneros 4, et al, Alfredo A Sadun 13 Brain. 2016 Mar;139(Pt 3):e17. doi: 10.1093/brain/awv339. Epub 2015 Dec 10.
Parsing the differences in affected with LHON: genetic versus environmental triggers of disease conversion)
Childhood-onset LHON accounts for approximately 8-10% of the total LHON affected; it can be considered an atypical variant of the disease. It is defined by the onset of symptoms before the age of 12 and can present as acute bilateral LHON, slowly progressive or insidious LHON, and in some cases subclinical/acute unilateral LHON. In unilateral cases, visual loss occurs in one eye, even in early infancy, while the fellow eye remains unaffected for variable periods ranging from weeks to several years, even till a later age (> 15 years old). This specific pattern, classified as insidious unilateral onset, could be due to subtle anatomical differences between the two eyes. [4,5,6] True long-term unilateral cases are rare but have been documented [7-9]
Although the distribution of genetic mutations does not differ from that observed in adults, childhood-onset LHON generally carries a more favourable prognosis, particularly in cases where symptom onset occurs before the age of 9. Visual recovery has been reported in 64.8–83.3% of eyes, with a final visual acuity of at least 0.5 achieved in 40.7–51.8% of eyes. [4,5]
It should be noted, however, that diagnosis in children, especially during the early years of life, can be extremely challenging due to the difficulty in recognizing symptoms, the variability in disease expression, and the involvement of an immature visual system.
Misdiagnoses with other optic neuropathies or amblyopia is therefore common, particularly in cases that remain unilateral for many years. [1, 7-12]
In conclusion, this case illustrates an unusual disease course, with a typical onset in adulthood and an earlier episode during childhood that had been previously diagnosed as amblyopia. The progression was also atypical, showing a gradual enlargement of the scotoma and a thinning of the RNFL and GCL, extending beyond one year from onset (slowly progressive course). Despite a relatively good final visual outcome, the patient developed—or possibly unmasked—a psychotic disorder, possibly influenced by the psychological burden of coping with a visual impairment in a young male, including challenges related to uncertainty, shifting family dynamics, and particularly the maternal relationship.
ECM
Reference
- Newman N. Hereditary optic neuropathies. In: Miller NR, Biousse V, Newman NJ, Kerrison JB, eds. Walsh and Hoyt’s Clinical Neuro-Ophthalmology. Philadelphia: Lippincott Williams & Wilkins; 2005:465–501.
- Yu-Wai-Man P, Bateman DE, Hudson G, Griffiths PG, Chinnery PF. Leber hereditary optic neuropathy presenting in a 75-year-old man. J Neuroophthalmol 2008;28:155.
- Dagi LR, Rizzo JF, 3rd, Cestari DM. Leber hereditary optic neuropathy in an octogenarian. J Neuroophthalmol 2008;28:156.
- Barboni P, Savini G, Valentino ML, La Morgia C, Bellusci C, De Negri AM, Sadun F, Carta A, Carbonelli M, Sadun AA, Carelli V. Leber’s hereditary optic neuropathy with childhood onset. Invest Ophthalmol Vis Sci. 2006 Dec;47(12):5303-9. doi: 10.1167/iovs.06-0520. PMID: 17122117.
- Barboni P, La Morgia C, Cascavilla ML, Hong EH, Battista M, Majander A, Caporali L, Starace V, Amore G, Renzo AD, Carbonelli M, Nucci P, Jurkute N, Chen BS, Panebianco R, De Negri AM, Sadun F, Parisi V, Bandello F, Sadun AA, Carelli V, Yu-Wai-Man P. Childhood-Onset Leber Hereditary Optic Neuropathy-Clinical and Prognostic Insights. Am J Ophthalmol. 2023 May; 249:99-107. doi: 10.1016/j.ajo.2022.12.014.
- Tomita G, Nyman K, Raitta C, Kawamura M. Interocular asymmetry of optic disc size and its relevance to visual field loss in normal-tension glaucoma. Graefes Arch Clin Exp Oph- thalmol. 1994; 232:290–296.
- Nagai A, Nakamura M, Kusuhara S, Kanamori A, Negi A. Unilateral manifestation of Leber’s hereditary optic neuropathy after blunt ocular trauma. Jpn J Ophthalmol. 2005;49:65–67.
- Barboni P. Sadun AA. Atlas of LHON. MEDonline; 2019. Yu-Wai-Man P, Newman NJ, Carelli V, et al. Natural his- tory of patients with Leber hereditary optic neuropathy-results from the REALITY study. Eye (Lond). 2022; 36:818–826. Baron Nelson M, O’Neil SH, Wisnowski JL, et al. Matu- ration of brain microstructure and metabolism associates with increased capacity for self-regulation during the transition from childhood to adolescence. J Neurosci. 2019;39(42):8362–8375.
- Ohden KL, Tang PH, Lilley CC, Lee MS. Atypical Leber hereditary optic neuropathy: 18 year interval between eyes. J Neuroophthalmol. 2016;36:304.
- Nikoskelainen EK, Huoponen K, Juvonen V, Lamminen T, Num- melin K, Savontaus ML. Ophthalmologic findings in Leber hereditary optic neuropathy, with special reference to mtDNA mutations (published correction appears in Ophthalmology. 1996;103:998). Ophthalmology. 1996;103:504–514.
- Riordan-Eva P, Sanders MD, Govan GG, Sweeney MG, Da Costa J, Harding AE. The clinical features of Leber’s hereditary optic neu ropathy defined by the presence of a pathogenic mitochondrial DNA mutation. Brain. 1995;118:319–337.
- Newman NJ. Leber’s hereditary optic neuropathy: new genetic considerations. Arch Neurol. 1993;50:540–548.